同日兩篇NEJM:爲漸凍症病人"解凍",基因療法臨牀試驗帶來治癒曙光
來源:學術經緯
多年前,冰桶挑戰讓“漸凍症”(肌萎縮側索硬化症,ALS)這種致命的疾病走進公衆的視野。大家熟悉的霍金教授,以及知名紀實作品《相約星期二》中的莫里·施瓦茨教授,都在漫長歲月中與ALS勇敢鬥爭。
我們知道,現有治療並不能治癒ALS,但科學家們正朝着這一目標不斷進行前沿探索和研究。《新英格蘭醫學雜誌》(NEJM)最新同期發表的2項重要研究,爲ALS患者帶來了一絲治癒的曙光:基因療法有效抑制了致病基因,改善了與疾病相關的指標!

圖片來源:123RF
在介紹兩項研究前,我們先簡要了解下ALS。ALS是一種主要累及運動神經元的神經系統變性疾病,患者通常會在發病後5年內因呼吸衰竭而死亡。遺傳風險佔到了發病影響因素的50%。
1993年,麻省大學醫學院的Robert H。 Brown教授及其合作團隊發現了首個與家族性ALS相關的基因——編碼SOD1(超氧化物歧化酶1)蛋白的基因。後續研究提示,錯誤摺疊的SOD1蛋白可能導致運動神經元出現異常,促進ALS發病;對於具有相應致病突變的ALS患者而言,調節SOD1突變基因的表達、降低毒性蛋白的水平可能具有治療作用。在動物模型中,這類療法減緩甚至逆轉了運動神經元的死亡。
這次,兩支研究團隊分別通過兩種基因療法,在攜帶SOD1突變ALS患者中探索了抑制SOD1基因表達的潛在治療益處。Brown教授表示:“自1993年以來我們一直致力於抑制SOD1基因,這些早期結果令人感到鼓舞。”

圖片來源:New England Journal of Medicine
第一項研究是由華盛頓大學神經病學學者與渤健公司研究團隊領銜的1/2期臨牀試驗。試驗藥物tofersen是一種反義寡核苷酸,可以通過調節SOD1信使RNA的降解,從而減少SOD1蛋白的合成。共48例受試者3:1隨機分組,接受4種不同劑量(20 mg/40 mg/60 mg/100 mg)的tofersen或安慰劑,給藥方式爲腰椎鞘內注射,治療爲期12周,每人共5劑藥物。
治療第85天的數據顯示,按劑量遞增,不同劑量tofersen組患者腦脊液中的SOD1蛋白濃度分別降低了2%、25%、19%和33%。最高劑量tofersen組的下降程度最明顯,這提示藥物確實對治療靶標發揮了作用。同時,接受tofersen的患者腦脊液中的神經絲濃度也降低了,這間接表明藥物減少了神經變性。
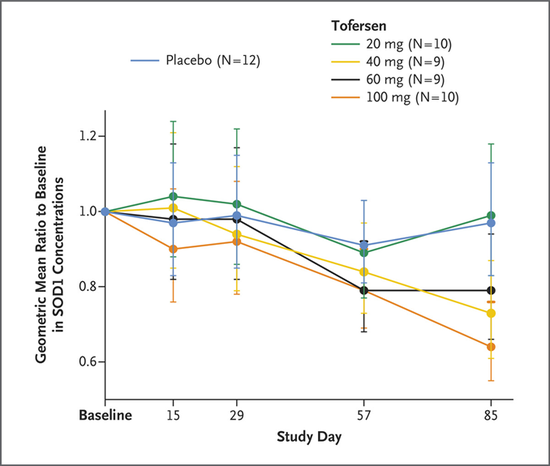
患者腦脊液內的SOD1蛋白總濃度相對於基線的變化
進一步分析顯示,SOD1基因突變與疾病快速進展有關的患者,潛在獲益更多。這些患者的腦脊液中神經絲濃度下降更多,患者功能量表評分下降更慢(提示功能衰退更慢)。
目前,一項3期、隨機、雙盲、安慰劑對照試驗及其長期擴展研究正在進一步評估tofersen的安全性和療效。

圖片來源:New England Journal of Medicine
第二項研究由麻省大學醫學院、麻省總醫院和哈佛醫學院團隊合作開展,是一項概念驗證研究。試驗藥物是一種腺相關病毒(AAV),這種AAV可以編碼靶向SOD1的微RNA。
在自然界中,微RNA可以破壞基因的RNA模板,阻止細胞將RNA進一步翻譯爲蛋白質。這項試驗中,研究團隊便是利用這種細胞機制,抑制SOD1基因和相應蛋白。值得一提的是,SOD1基因有180多種不同突變與ALS相關。研究團隊確定了這些突變DNA序列的共同點,開發了能夠靶向絕大多數SOD1基因突變的相對通用的AAV療法。
當這種AAV被注射進入脊髓液後,能夠在整個脊髓組織中遞送靶向SOD1的微RNA,進而減少脊髓組織中SOD1蛋白的產生。
試驗中,2名患有家族性ALS的受試者接受了單次鞘內注射AAV。
患者1治療後15.6個月(ALS發病後20.5個月)時死於呼吸衰竭。儘管最終未能拯救患者生命,但患者的右腿力量曾有過暫時性的改善,此外,屍檢時結果顯示,相較於其他未接受治療的SOD1突變ALS患者以及健康對照人羣的水平,其脊髓組織中的SOD1蛋白水平更低。

患者1屍檢的腰骶髓樣本組織學分析顯示,與左側一半脊髓(左圖)相比,右側一半脊髓(右圖)保留的運動神經元相對更多
患者2的腦脊液SOD1蛋白水平未受影響,但ALS功能綜合指標評分和肺活量在12個月內都保持穩定。
在安全性方面,患者1接受治療後出現了腦膜神經根炎;患者2預先接受了免疫抑制藥物,未出現炎症反應。
整體來說,研究團隊認爲,初步試驗表明,微RNA鞘內給藥有潛力治療SOD1相關ALS。接下來,研究團隊計劃在安慰劑對照試驗中測試第二代這種臨牀候選藥物的療效。

圖片來源:New England Journal of Medicine
在NEJM同期刊發的社論文章指出,雖然仍需更多研究和證據積累,這兩項研究表明,精準醫療有可能治療單基因突變相關的神經退行性疾病。同時,針對其他基因突變相關ALS的療法也正在蓬勃發展,處於早期臨牀或臨牀前階段。包括將反義寡核苷酸用於與C9orf72突變或TDP43蛋白表達相關的ALS等疾病。
“這些進展標誌着ALS治療的新開端,其中某些亞型有望得到治癒。通過從具有特定遺傳特徵的患者亞組開始,科研人員們正在爲具有這種致命疾病遺傳風險的患者帶來新的希望。”
期待這些療法能在臨牀試驗中得到更多驗證,爲ALS患者帶來“解凍”希望!